Here's hoping the bacteria don't crawl off the screen
Apr 12, 2010, 5:13p - Science
(This is the third in a 3-part series on the biology of bacterial chemotaxis. The first post described some basic ideas about biological analysis, and the second post introduced bacteria and their chemotactic behavior. And don't forget the bonus video post.)
Alright, now that the context has been set, I can finally dive into my bacterial chemotaxis simulator, which is what motivated this whole series of posts to begin with (which has taken far too many months to publish).
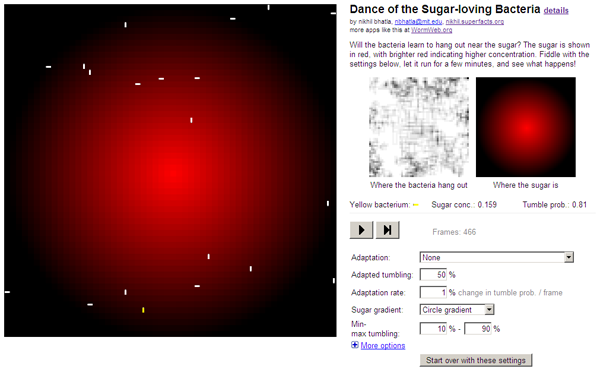
What is it?
This is an interactive simulator of bacterial chemotaxis that I built in October 2008. It's "interactive" because you can change a bunch of settings and see how the virtual bacteria change their behavior. On the left side is the bacterial playground, where the bacteria (small white rods) swim around. Red spots indicate there's a chemoattractant there (say, sugar), and the brighter the red, the higher the sugar concentration. One of the bacteria is colored yellow (it's hard to see when the simulation is moving fast, but it's there), and that bacterium's current tumble probability and sugar concentration are shown on the right as it moves through the environment.
Why did I build this?
I built this simulator because I wanted to get a better understanding of how bacterial chemotaxis might work. The simulator has a bunch of bacteria swimming around in a world of different sugar concentrations, and I wanted to see what it would take to get the bacteria to properly chemotax, to swim to the highest sugar concentration and spend the most time there. As I mentioned before, I really believe that a good test of whether we understand something is whether we can build a simulation of that very thing. So building this simulator was my quick test for whether we (as a scientific collective) understand how bacterial chemotaxis actually works. Of course, I'm not even close to being an expert on chemotaxis, but I figured I'd give it a quick 'n dirty shot. Plus, I like writing web-apps.
When I first started making this simulator, I had the impression that a bacterium moves to the highest sugar concentration simply by tumbling less if the current sugar levels are high, and tumbling more if the current sugar levels are low. As I'll show below, this simple strategy is insufficient for chemotaxis (at least in my simulator given the various things I've tried). What I've found instead is that it's critical for the bacterium to keep a memory of the recent sugar concentrations it's experienced. To chemotax, the bacterium must change its tumble probability not only if sugar levels are changing, but also if sugar levels are staying the same. If the sugar levels are relatively constant, it needs to return to a basal tumbling probability. If it doesn't, it will swim away from areas of high concentration. This may be obvious in retrospect, but I found it quite surprising and unexpected. Detailed results of experiments with the simulator are described in the "What have I learned" section below.
How does it work?
As mentioned before, bacteria can either move in "runs" or they can tumble. A run means that a bacterium just moves straight in the direction it was going, while a tumble means that it randomly spins around and changes the direction that it's moving in. In this simulation, each bacterium gets a turn to move. On each turn, it randomly chooses whether to move forward one square, or to randomly choose a new direction to move in and then moves one square in that direction. At the end of its turn the bacterium updates its tumble probability, depending on the sugar concentration at the new location (and perhaps its sugar history, as described below).
So let me run through an example of precisely how a turn works. Let's say it's a bacterium's turn to move, and it's current tumble probability is 0.9. This is a very high probability, and says that there's a 90% chance that the bacterium will tumble on its turn. The program then picks a (pseudo) random number uniformly distributed between 0 and 1, and if that number is less than the tumble probability of 0.9, it will tumble. If it's greater than 0.9, it will just move forward. Let's say it draws a 0.4. So now it's going to tumble. Now, it draws a second random number to decide which direction to tumble in (in this simulator a bacterium is limited to just moving in 4 directions - up, down, left, and right). A bacterium has an equal probability of tumbling into any of the 4 directions. So let's say it draws a 0.2, which means it will move left. It then moves one spot to the left.
Now here comes the interesting part. Now that the bacterium has moved, it's time to update its tumble probability for the next turn. So what seems like a reasonable, simple formula for doing this? Let's imagine that if the sugar concentration is low, that means the bacterium isn't moving in a good direction, so maybe it should change its direction. And if the sugar concentration is high, the bacterium is in a good place, and should just keep swimming forward. It could just stop swimming, but maybe there's even more sugar if it keeps going the way its been going. One way to implement this sort of algorithm is to just set the tumble probability to 1 minus the current sugar concentration (which is limited to values between 0 and 1). So if the bacterium was at a spot with a sugar concentration of 0.13, it would update its tumble probability to 1 - 0.13 = 0.87. Conversely, if the sugar concentration were high at 0.9, it would have a tumble probability of 0.1.
This is the default update formula, and is specified when the Adaptation field is set to "None". Before I talk too much about what the Adaptation field is, let me explain the other parts of the simulator (on the right side, from top to bottom):
- Where the bacteria hang out: This is a location "heat map" of where the bacteria hang out, with black shading indicating that bacteria spend more time there than spots with whiter shades. This is the ultimate test of whether the bacteria are successfully chemotaxing. If the heat map is nearly uniformly grey, the bactera hang out everywhere equally. If the heat map is dark in the center when the bacteria are exploring a circle gradient, that means that they're successfully chemotaxing.
- Where the sugar is: This is a visual representation of where the sugar is in the bacteria's environment. As I mentioned above, brighter red indicates higher sugar concentrations. The default is a circle gradient, with the highest concentration in the center of the circle.
- A row describing the current sugar concentration and tumble probability of the yellow bacterium.
- Pause/Play button: Pauses the simulation when it's running, and plays the simulation if it's paused.
- Next frame button: Runs only one frame (turn) of the simulation, pausing immediately after that turn is finished. You can use this to step through the simulation to watch individual bacteria, since it updates a bit too fast otherwise.
- Frames: How many frames (turns) the simulation has run so far.
These next settings actually affect whether the bacteria successfully chemotax. So this is the stuff you should fiddle with to see how the bacteria's behavior changes.
- Adaptation: This sets whether the bacteria adapt to the sugar they experience, and if so, how quickly they adapt. What do I mean by adaptation? I mean that a bacterium will "get used to" the current sugar concentration and readjust its tumble probability to a "normal" level, even if the sugar concentration isn't changing. This is just like how your eyes readjust so you can see in a darkened room. There are 3 options for this setting:
- None: The default setting, this specifies that the bacteria don't adapt at all, and the tumble probability is just updated to 1 minus the current sugar concentration on each turn (as described above).
- Tumble returns to steady-state (linear): When set to this option, a bacterium's tumble probability will update to approach the value of the adaptated tumbling field, minus the difference in current versus previous sugar concentrations. How much it will approach is specified by the adaptation rate field. As an example, let's say that a bacterium's tumble probability is currently 0.6, it's adaptation rate is 1% (a probability of 0.01), the adapted tumbling probability is 50%, the current sugar concentration is 0.2, and the previous concentration was 0.3. So the new tumble probability will be (0.6 - 0.01) - (0.2 - 0.3) = 0.69. This formula may seem a bit complicated, but notice that it takes into account 2 important things: an adjustment to adapt back to a baseline probability, and the fact that the tumble probability increases if the sugar concentration decreases, and decreases if the sugar concentration increases. Now that I write this out, it seems sort of complicated, but hopefully it makes a little bit of sense.
- No tumble up, tumble otherwise (2 state): This is an update algorithm inspired from a paper by the van Oudenaarden lab at MIT (see citation at the end). Basically, a bacterium's tumble probability is set to 0 if the current sugar concentration is higher than the average sugar concentration from the last 3 turns. If it's lower than the average from the last 3 turns, the tumble probability is set to the adapted tumbling probability. The idea here is the same: if sugar levels are increasing, never tumble, but if they are decreasing or staying the same, tumble like crazy. Note that if this adaptation algorithm is used, the min-max tumbling setting below is ignored.
- Adapted tumbling: The tumble probability that is used in the adaptation algorithms described above. If adaptation is set to "None", this field has no effect. The default value is 50%.
- Adaptation rate: The rate at which a bacterium's tumble probability relaxes to the adapted tumbling probability. This only affects the simulation if adaptation is set to "Tumble returns to steady-state (linear)". The default value is 1%, meaning that if the current sugar concentration equals the previous sugar concentration, the tumble probability will be updated to be 1% closer to the adapted tumbling probability. So if the current probability is 0.6, and the adapted probability is 0.5, the new probability will be 0.59. It will get 1% closer on each turn until it reaches 50%, assuming the sugar concentration remains unchanged.
- Sugar gradient: This is the shape of the sugar gradient. The options are:
 |  |  |
| Left/right | Circle | Circle gradient (default) |
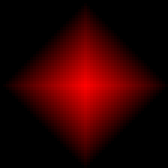 |  | |
| Diamond gradient | Corner gradient | |
- Min-max tumbling: The first value specifies the minimum tumble probability for a bacterium, and the second value sets the maximum. Think of this as a floor and ceiling for a bacterium's tumble probability, regardless of how the tumble probability is updated (which you can set with the adaptation settings below).
In addition to the options above, there are some extra options if you click on the "More options" link:
- Room size: This sets the size of the bacteria's environment, which must be square. The default value is 56 spaces square.
- Bacteria number: This sets the number of bacteria to simulate. The default value is 20 bacteria.
- Bacteria speed: This sets the update rate for the simulation. The default value is 1000 turns per second per bacteria. That means that, assuming your browser and CPU can handle it, the simulation will run 1000 turns for each bacteria in 1 second.
- Hang-out updating: This sets how many turns have to pass before the "Where the bacteria hang out" heat map is updated. The default value is to update the heat map after 100 turns.
- Auto-stop after: This tells the simulator when to automatically stop running the simulation. Without this setting the simulation would just run as long as your browser window is left open. I added this option because it gives you a common stopping point at which to compare simulation runs with different option values. The default value is to stop after 100,000 turns for each of the bacteria.
So that's a comprehensive list of the various options available in the chemotaxis simulator.
So what have I learned from this?
Basically, I learned that adaptation is critical for bacterial chemotaxis. Adaptation can be thought of simply as using a "memory" of a previous time to alter current behavior, by comparing then with now. In fancy lingo, I'd say that adaptation is just a "temporal comparator". By keeping a memory of recent sugar concentrations, the bacteria are able to chemotax to the peak of the sugar gradient. Without this memory, with just the current sugar concentration, the bacteria cannot chemotax (at least in this simulation under the conditions I tested).
Here are the heat map results showing where the bacteria hang out in 2 different worlds using 3 different adaptation algorithms. Each heat map is the result of a single simulation which includes: 20,000 frames of simulation, 20 bacteria, 56x56 world size, 10-90% min-max tumbling, 50% adapted tumbling, and 1% adaptation rate.
| Sugar gradient: |
|
|
| No adaptation: |
 |
 |
| Linear adaptation: |
 |
 |
| 2-state adaptation: |
 |
 |
As you can see above, when there isn't any adaptation, the bacteria hang out all over the place, so the heat maps look like a formless grey blur. If you actually watch the bacteria moving around, you can see that at low concentrations, the bacteria just tumble like crazy and find it difficult to leave because they're always tumbling. But at high concentrations the bacteria just sail straight through, eventually ending up back in low concentration territory, where they begin to tumble like crazy again.
With linear adaptation, you can start to see the shape of the sugar gradient in the heat maps of where the bacteria hang out. This means that the bacteria are chemotaxing somewhat. Finally, with 2-state adaptation, you get the clearest picture of the sugar gradient from the heat maps, indicating that the bacteria are efficiently chemotaxing and staying in the areas of high sugar concentration.
Alright, that's pretty much it folks. I have various caveats listed below, but if you've made it this far, you may just want to play around with the simulator some more. Hopefully I haven't been too verbose, as I optimized this post for for completeness and clarity over pithiness.
Caveats and other notes
- Bacteria only move in 4 directions, not all 8 (or more).
- My simulated bacteria seem to tumble too much, compared with the video in the previous post.
- The real bacteria from the video seem to have a curvature to their paths, which the simulated bacteria don't have (they're runs are in straight lines).
- Remember, this is just a simulator. I don't mean to draw strong conclusions about biology based on this web-app; it's more meant to be a fun toy that can be played around with, and that may also inspire novel non-virtual experiments.
- It's worth noting that many other people have already made bacterial chemotaxis simulators. Many of these have been published in the academic literature. Many of them are more complex and more accurate than what I've built. But what's unique about mine is that it is a webapp that you can run for yourself in a web browser. You can tweak variables and tinker with things and see how the behavior of the simulated bacteria change. So I think it's pretty nifty.
- One significant limitation of my simulator is that it models bacteria at a behavioral level, calculating the tumble probability as a function of the changing sugar concentration. After making this simulator, I really wanted to model bacteria at a molecular level, simulating the protein-protein interactions that actually cause the changes in tumble probability. Comparing such a simulation to real bacteria would be an excellent way to measure how much we really understand about how bacterial chemotaxis works. However, it's been a year since I made this simulator, and realistically I'm not going to make the molecular simulator, so I decided to blog about what I had so far.
I hope you enjoyed this web-app, and maybe even learned a little bit!
--
References:
- Mittal..van Oudenaarden 2003. "Motility of Escherichia coli cells in clusters formed by chemotactic aggregation." PNAS. PDF
Read comments (2) - Comment
becca
- Apr 12, 2010, 7:06p
Yay! Es un simulador muy bueno.
Rolf
- Aug 25, 2010, 5:04a
Nice app! I've programmed a very simple optimization algorhitm in Matlab and SciLab based on bacteria behavior.
My bacteria takes a step of constant length in its direction. Then, it compares 'sugar concentration'. If lower, it tumbles, otherwise its orientation stays the same.
Interestingly, this procedure finds the minimum of the Rosenbrock function and other test functions.
« Bacteria don't have to be icky
-
Video of real bacterial chemotaxis »
|